Split FastQ and FastA files
Introduction
This tool splits FastQ and FastA files into multiple output files using SeqKit, a high-performance toolkit implemented in Go. It supports several splitting options, including dividing a file into equal parts, splitting by a defined number of sequences, grouping sequences by their IDs, or extracting specific sequences into a new file. The tool is located in the General Module of OmicsBox under FastQ Tools → Split FastQ/FastA files with SeqKit.
For more information about SeqKit, please visit: Shen W, Le S, Li Y, Hu F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS One. 2016 Oct 5;11(10):e0163962. doi: 10.1371/journal.pone.0163962
Inputs
It is possible to provide multiple files at once to apply the same splitting operation to all of them, including a mix of FastQ and FastA files. For each input, it is necessary to specify whether it is single-end or paired-end. The paired-end option is only available for FastQ files, and the suffix of both files must match the pattern described below.
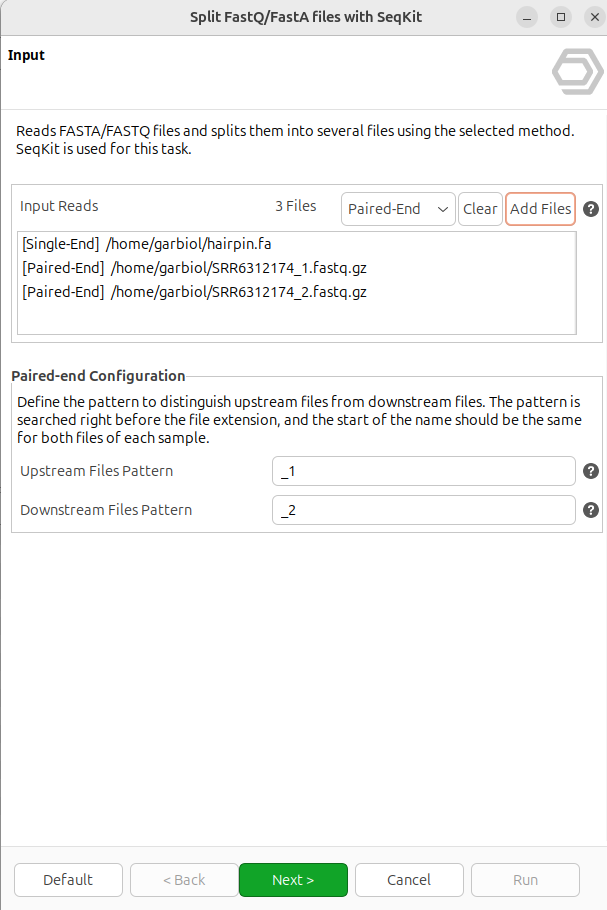
Figure 1. Input page
Splitting options
This tool supports four different splitting modes.
Splitting by ID is only allowed for FastA files. These options will be disabled if any FASTQ file is selected.
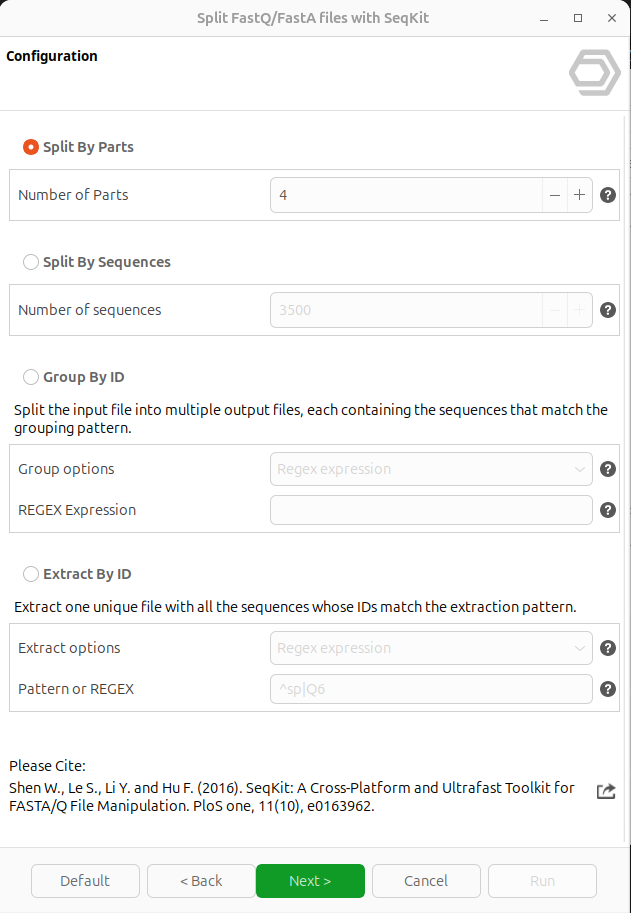
Figure 2. Splitting options page
- Split By Parts: Splits the input files into N equal parts. The N is the total number of parts and is defined by the "Number of Parts" parameter.
- Split By Sequences: Splits the input files into multiple parts with at most N sequences each. The N is the maximum number of sequences per output file and is defined by the "Number of Sequences" parameter.
-
Group By ID: Split each input file into multiple output files, each containing all the sequences whose ID matches the grouping pattern. The grouping pattern is defined using regular expressions, and there are 4 predefined grouping patterns:
- First word: Groups all the sequences that share the same first word in their ID. The first word includes everything up to the first whitespace character (space, tab, newline, etc.). The regular expression is "^(\S+)"
- Until - or _ : Groups all sequences that share the same characters at the beginning of their ID, until a dash: " - " or an underscore: " _ " is reached. The regular expression is "^([^_-]+)"
- Species name: Groups all the sequences belonging to the same species, based on matches to the pattern <genus_species> at the beginning of the id.
The regular expression is: "^([A-Z][a-z]+_[a-z]+)" - UniProt Accession: Groups all the sequences with the same UniProt Accession (its position in the ID does not matter, but is expected to follow the UniProt format). The regular expression is "^\w+|([^|]+)|"
sp|Q6GZX4|001R_FRG3G
TACACTCTTTCCCTACACGACGCTCTTCCGATCT
sp|Q6GZX3|002L_FRG3G
MSIIGATRLQNDKSDTYSMSIIGATRLQNDKSDT
sp|Q6GZX4|002R_IIV3
DVGTQNILRDLVNLPVEMSGDLQVMAYTKDDVGT-
In this example, when grouping by UniProt Accesion, the first and third sequences are written to the same output file, as they share the same UniProt Accession: 'Q6GZX4'. The second sequence is written to a different output file.
-
Regex expression: Groups the sequences by the regular expression defined by the user.
Take into consideration that every sequence with a different match of the defined pattern results in the creation of a new output file, so very specific expressions like ^(SPECIFIC_NAME) are not recommended and are more suitable for the 'Extract By ID' option.
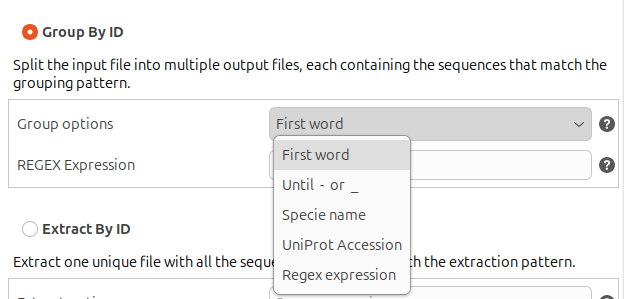
Figure 3. Grouping options
- Extract By ID: Extract a single file containing all the sequences whose ID matches the extraction pattern. The extraction pattern is defined using regular expressions, and there are 2 predefined extraction patterns:
- Starts with: Extracts only the sequences whose ID starts with the text provided by the user. It is recommended to avoid special characters like ., *, +, [, ], (, ).
The regular expression is ^<user_text> - Contains: Extracts only the sequences whose ID contains (in any position) the text provided by the user. It is recommended to avoid special characters like ., *, +, [, ], (, ).
The regular expression is <user_text> - Regex expression: Extracts only the sequences that match the regular expression defined by the user.
- Starts with: Extracts only the sequences whose ID starts with the text provided by the user. It is recommended to avoid special characters like ., *, +, [, ], (, ).
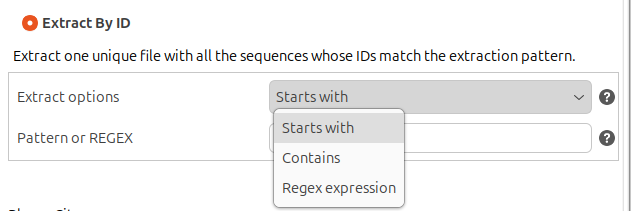
Figure 4. Extracting options
Output
The resulting files are saved in the directory specified by the user. They are automatically named using the input file name plus a distinctive numeric suffix (E.g. hairpin.part_001.fa & hairpin.part_002.fa).
If the input file is split by its ID, the resulting files use the distinctive ID pattern as a suffix (Ex: hairpin.part_hsa.fa, hairpin.part_cal.fa, hairpin.part_mmu.fa...)
OmicsBox Engine
This tool can be run from the command line via the OmicsBox Engine.
Command: omicsbox fasta-splitter [options]
Inputs
| Flag | Type | Required | Description |
|---|---|---|---|
--i-input-files-single-end |
file (multiple) | No | Single-End |
--i-input-files-paired-end |
file (multiple) | No | Paired-End |
Parameters
| Flag | Type | Default | Range / Candidates | Required | Description |
|---|---|---|---|---|---|
--upstream-pattern |
string | _1 | No | Upstream Files Pattern | |
--downstream-pattern |
string | _2 | No | Downstream Files Pattern | |
--split-value-parts |
integer | 2 | ≥ 2 | No | Number of Parts |
--split-value-sequences |
integer | 10000 | ≥ 1 | No | Number of Sequences |
--group-by-options |
enum | regex | firstWorduntilSignspecieNameuniprotregex |
No | Group Options |
--group-value-id |
string | No | REGEX Expression | ||
--extract-by-options |
enum | regex | startscontainsregex |
No | Extract Options |
--extract-value-id |
string | No | Pattern or REGEX | ||
--filter |
enum | parts | partssequencesgroupIdextractId |
No |
Parameter relationships
| Flag | When | Effect | Affected flags |
|---|---|---|---|
--filter |
sequences |
disables | --split-value-parts |
--filter |
sequences |
enables | --split-value-sequences |
--filter |
groupId |
disables | --split-value-parts |
--filter |
groupId |
enables | --group-by-options, --group-value-id |
--filter |
extractId |
disables | --split-value-parts |
--filter |
extractId |
enables | --extract-by-options, --extract-value-id |
Note:
--upstream-pattern,--downstream-patternare context-dependent — they apply, and their valid values are determined, only for certain input configurations.Global options (
--local-folder,--cloud-folder,--output-format,--config,--detach,--verbose, …) are shared by every Engine tool and are not repeated here — see the OmicsBox Engine reference.