Functional Annotation with EggNOG Mapper
Introduction
EggNOG Mapper annotates novel sequences (genes or proteins) using precomputed eggNOG orthology assignments. Typical uses include annotating genomes, transcriptomes, and metagenomic gene catalogs. Orthology-based transfer is often more specific than simple homology, because annotations are not taken from paralogs that may have diverged in function.
The methodology and the underlying database are described on the eggNOG methods page.
EggNOG Mapper produces its own results object, separate from a functional annotation project: a table where each row is a query sequence with the functional information transferred from its eggNOG orthologs. These annotations can later be merged into a functional annotation project (see Merge EggNOG GOs to a Functional Annotation Project).
Run
EggNOG Mapper can be launched from:
- functional analysis → Functional Annotation with EggNOG Mapper.
- The Side Panel of a Functional Annotation Project, in the EggNOG group.
When the job is started from a loaded project, the sequences come from the project and the Input page is skipped. When it is started from the menu without a project, the input files are selected on the Input page first.
Input
The Input page is shown only when no project is loaded.
-
Genes or Proteins: One or more multi-FASTA files containing gene or protein sequences (extensions such as
.fasta,.faa,.fna,.fa,.fn,.ffn). -
Input Type: How sequences are searched against eggNOG (default CDS):
-
Proteins: Amino acid sequences are searched with BLASTp.
- CDS: Coding sequences are translated to proteins, then searched with BLASTp.
- Genes/Contigs: Nucleotide sequences are searched in all six reading frames with BLASTx.
OmicsBox can detect whether files contain protein or nucleotide sequences and warns if the chosen Input Type does not match. Sequences longer than 2.5 Mb are filtered automatically and skipped.
Configuration
The Configuration page sets the search and annotation options (Figure 1).
Search filters
- Minimum Hit e-Value: Minimum e-value for seed ortholog hits in the homology search phase (default 1.0E-3). Lower values are more stringent; higher values are more permissive.
Annotation options
-
Taxonomic Scope: Restrict annotation to orthologs from a chosen clade. Default Adjust Automatically picks the best scope per query sequence. Select a specific taxon when the origin of the data is known.
-
Target Orthologs: Which ortholog relationships are used to transfer function (default All):
-
All: All ortholog types (recommended).
- One to One: Single ortholog per pair of species (most stringent).
- One to Many: One-to-one and one-to-many orthologs.
- Many to One: One-to-one and many-to-one orthologs.
-
Many to Many: All types including many-to-many (least stringent, most comprehensive).
-
GO Evidence: Which Gene Ontology (GO) terms are transferred (default Non-Electronic):
-
Experimental: Only terms with experimental evidence codes (fewer, high-confidence terms).
- Non-Electronic: All evidence types except electronic inference only (recommended balance of quality and coverage).
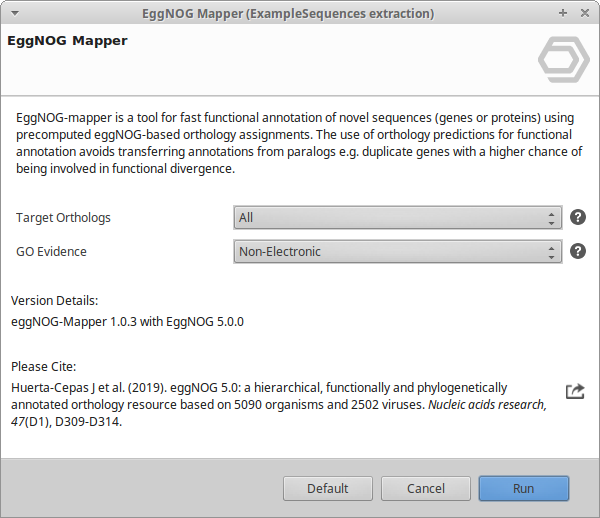
Figure 1. EggNOG Mapper wizard, Configuration page.
Results
The result is an EggNOG Mapper results object: a table with one row per query sequence holding the functional information transferred from its eggNOG orthologs (Figure 2). This is a standalone object, not a functional annotation project; its GO terms and enzyme codes can be added to a project afterwards (see Merge EggNOG GOs to a Functional Annotation Project). The run also produces a Summary Report with totals for GO terms, COG categories, and orthologous groups.
Results table
The table can be sorted and filtered. The columns shown by default are:
- Type. The orthologous group type the annotation is transferred from: COG (prokaryotic Clusters of Orthologous Groups), KOG (eukaryotic orthologous groups), or ENOG (eggNOG non-supervised orthologous groups).
- Query ID. The query sequence identifier.
- Gene Name. The predicted preferred gene name.
- EggNOG Description. The functional description of the matched orthologous group.
- E-Value. The e-value of the seed ortholog hit.
- Bit-Score. The bit-score of the seed ortholog hit.
- Best Tax-Level. The taxonomic level of the orthologous group from which the annotation was transferred.
- EC Codes. The Enzyme Commission numbers assigned.
- #GO. The number of GO terms transferred.
- GOs. The GO term identifiers.
- GO Names. The names of the GO terms.
- KEGG KO. The KEGG Orthology (KO) identifiers.
- KEGG Pathway. The KEGG pathways.
The following columns are hidden by default and can be shown from the column selector:
- EggNOG Protein. The seed ortholog protein matched in eggNOG.
- Taxa Scope. The taxonomic scope used for the annotation.
- KEGG Module, KEGG Reaction, KEGG RCLASS, and KEGG TC. Additional KEGG cross-references: modules, reactions, reaction classes, and transporter classification.
- Brite. KEGG BRITE functional hierarchies.
- CAZy. Carbohydrate-Active enZymes families.
- BiGG. BiGG metabolic reaction identifiers.
- Matching OGs. The orthologous groups matched by the query.
- COG Categories. The COG functional category classification.
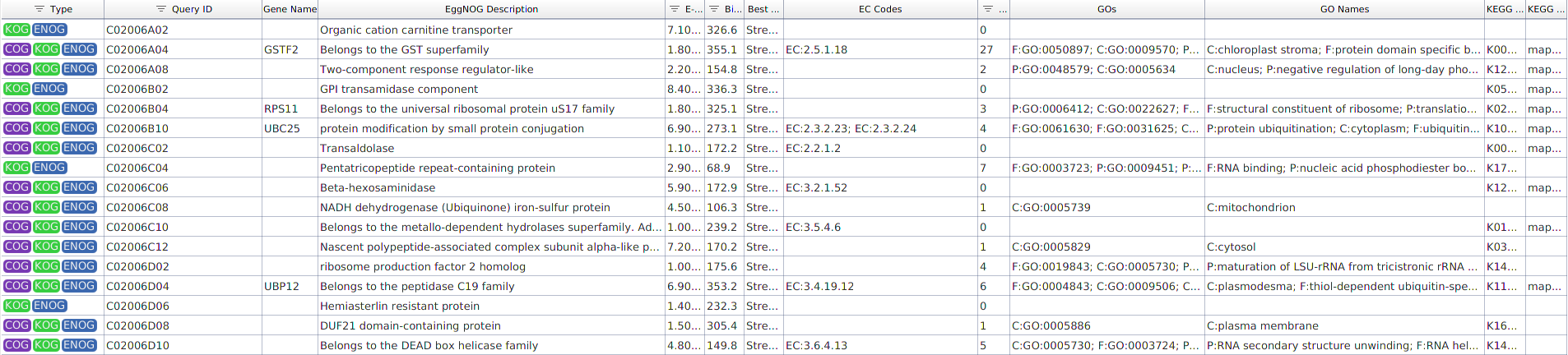
Figure 2. EggNOG Mapper results table.
Side Panel
Actions
- Fisher's Exact Test. Run an over-representation enrichment analysis on the EggNOG annotations. See Fisher's Exact Test.
- GO Slim. Reduce the GO annotations to a GO-Slim subset. See GO-Slim & Remove GO-Slim.
Export
- Export Table. Export the results table to a tab-separated text file.
Context Menu
Right-click a row to open its context menu. Apart from the generic Context Menu options, the EggNOG results have:
- Show GOs. Open the GO terms assigned to the selected sequence.
- Show Annotation Details. Open a detailed report of the eggNOG annotation for the sequence, including link-outs and GO information (Figure 3).
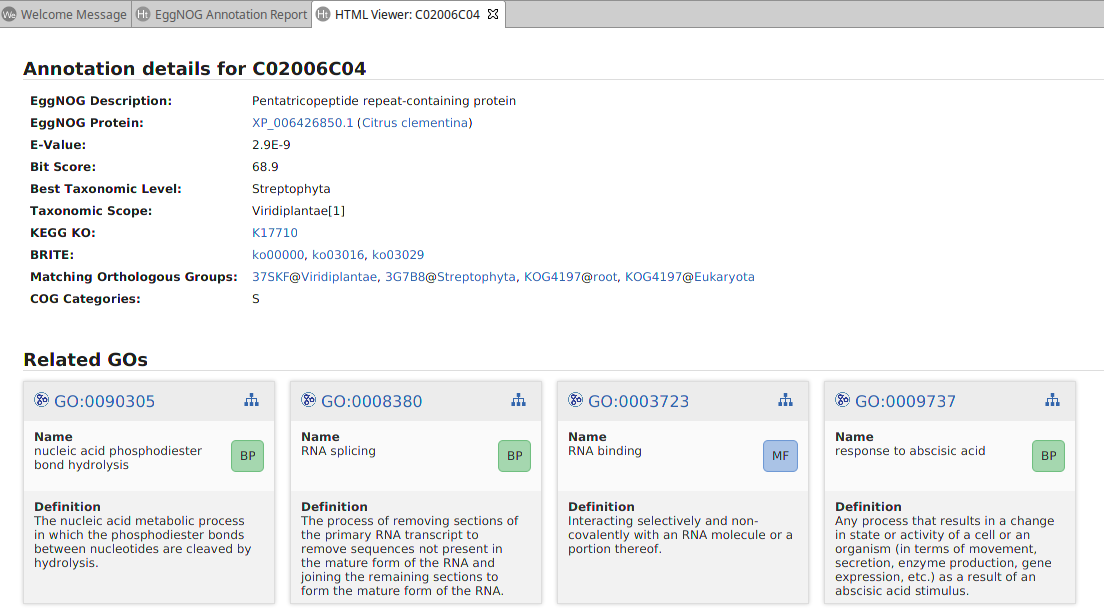
Figure 3. EggNOG Mapper annotation details.
Merge EggNOG GOs to a Functional Annotation Project
The EggNOG Mapper results are a standalone object. To incorporate their annotations into a functional annotation project, open the project and, in its Side Panel → EggNOG → Merge EggNOG GOs, select the EggNOG Mapper results to merge.
The GO terms and enzyme codes (EC) from the EggNOG results are added to the project's annotations; when a sequence already has annotations, the new ones are added to the existing set. The annotations to merge can be filtered by e-value or bit-score. When the merge finishes, a bar chart reports the total number of GO terms and enzyme codes added to the project.
OmicsBox Engine
This tool can be run from the command line via the OmicsBox Engine.
Command: omicsbox eggnog-mapper [options]
Inputs
| Flag | Type | Required | Description |
|---|---|---|---|
--i-sequences |
file (multiple) | No | Genes or Proteins |
--i-type-options |
enum | No | Input Type |
--i-local-project |
file | No | Sequence Project (.box) |
Parameters
| Flag | Type | Default | Range / Candidates | Required | Description |
|---|---|---|---|---|---|
--taxonomic-scope |
enum | auto | auto84992225057 |
No | Taxonomic Scope |
--target-orthologs |
enum | all | allone2onemany2oneone2manymany2many |
No | Target Orthologs |
--go-evidence |
enum | non-electronic | experimentalnon-electronic |
No | GO Evidence |
--blast-expect-value |
enum | 1.0E-3 | 1000105 |
No | Minimum Hit e-Value |
Global options (
--local-folder,--cloud-folder,--output-format,--config,--detach,--verbose, …) are shared by every Engine tool and are not repeated here — see the OmicsBox Engine reference.
References
Huerta-Cepas J et al. (2019). eggNOG 5.0: a hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Research, 47(D1), D309-D314.