Diamond Blast
Introduction
Diamond is an alternative to the official NCBI Blast software, developed for high-performance analysis of large sequence datasets. DIAMOND is a sequence aligner for protein and translated DNA searches. Key characteristics include:
- Protein and translated DNA pairwise alignment at speeds 100x to 10,000x faster than Blast.
- Alignments for frameshifts in long-read analyses.
This makes Diamond especially useful for larger datasets (5000+ query sequences). In OmicsBox, Diamond runs on the dedicated cloud infrastructure. DIAMOND is currently developed by Benjamin Buchfink at the Drost lab, Max Planck Institute for Biology, Tübingen, Germany.
Diamond Blast is one of the ways to run a Blast search in OmicsBox. See Blast Search for the overall Blast workflow and where to launch it.
Cloud Units consumption
Diamond runs on OmicsCloud and consumes Cloud Units, like the other cloud alignment tools of the Functional Analysis module (CloudBlast, CloudBlast Against Custom Database, and InterProScan). All subscriptions include a Cloud Units balance. Consumption is based on the CPU time used, which depends on the number and length of the query sequences, the selected database, the sensitivity mode, and the other parameters. The current balance and the consumption of each job can be monitored in View → Cloud Usage. For a full description of Cloud Units, see Cloud Computation and Storage.
To reduce Cloud Units consumption:
- Restrict the search with a taxonomy filter, or blast against a smaller database or an NR subset. The smaller the database, the more sequences can be analysed with a given number of Cloud Units.
- Use a lower sensitivity mode when only close homologs are needed; higher sensitivity modes scan more thoroughly and cost more CPU time.
Run
Select Diamond on the first page of the Blast wizard. The Diamond Blast Configuration page provides the following parameters:
- Blast Mode. The algorithm to use:
blastp: compares an amino acid query sequence against a protein sequence database.blastx: compares a nucleotide query sequence, translated in all reading frames, against a protein sequence database. Used to find potential translation products of an unknown nucleotide sequence.
- Sensitivity Mode. Controls the trade-off between speed and sensitivity of the alignment. Faster modes run quicker but miss distant homologs, while more sensitive modes detect increasingly remote matches at a higher computational cost. From fastest to most sensitive, the options are Fast (for hits above 90% identity), Mid Sensitive, Standard (Diamond's default, for hits above 60% identity), Sensitive (above 40% identity), More Sensitive (the OmicsBox default), and Very Sensitive (best sensitivity, including matches below 40% identity).
- Blast DB. The protein database to search in; several dated versions of three databases are available. As a rule of thumb, NR is the best option for a broad search, whereas SwissProt or RefSeq are more appropriate for a more specific annotation.
- NR (Non-Redundant). A collection of all publicly available protein sequences, including those from SwissProt and RefSeq. It is the most comprehensive option, but searches take longer and can return more false positives.
- RefSeq. NCBI's high-quality, expert-curated set of reference proteins with manual annotation. Smaller than NR, it increases the specificity of the search.
- SwissProt. The manually annotated, non-redundant, functionally characterized section of UniProtKB. It also offers high specificity and, being the smallest, the fastest searches, at the cost of lower coverage.
- Taxonomy Filter. Search for Blast results only in the selected taxonomy. This is highly recommended to obtain more specific results, and to considerably reduce computational costs. The taxonomy filter can be to the level of kingdom (e.g. Viridiplantaea, Fungi, Bacteria, etc), family, genus, or even species.
- Filter option. How the taxonomy list is used: Included blasts only against the sequences belonging to the specified taxonomies, while Excluded blasts against everything except them.
- Disable Taxonomy Filter. When enabled, no taxonomy filter is applied and the search runs against the full database. It should be enabled with caution.
- Blast Expectation Value (e-Value). The statistical significance threshold for reporting matches. Matches whose statistical significance is greater than the threshold are not reported. Lower thresholds are more stringent and lead to fewer chance matches.
- Optimize for Long Query Seqs. Improves performance for long query sequences of 30 kbp and longer. It is available only in
blastxmode, and enabling it disables the Number of Blast Hits option. - Number of Blast Hits. The number of alignments to report (0-100).
- HSP Length Cutoff. A cutoff for the minimal length of the first HSP of a Blast hit, used to exclude hits with only small local alignments. The length corresponds to amino acids or nucleotides depending on the Blast type.
- HSP-Hit Coverage. The minimum coverage between the hit sequence and its best HSP, as a percentage. For example, 80 means the aligned HSP must cover at least 80% of the length of the hit. It ensures that the hit is contained in the query by more than the specified percentage.
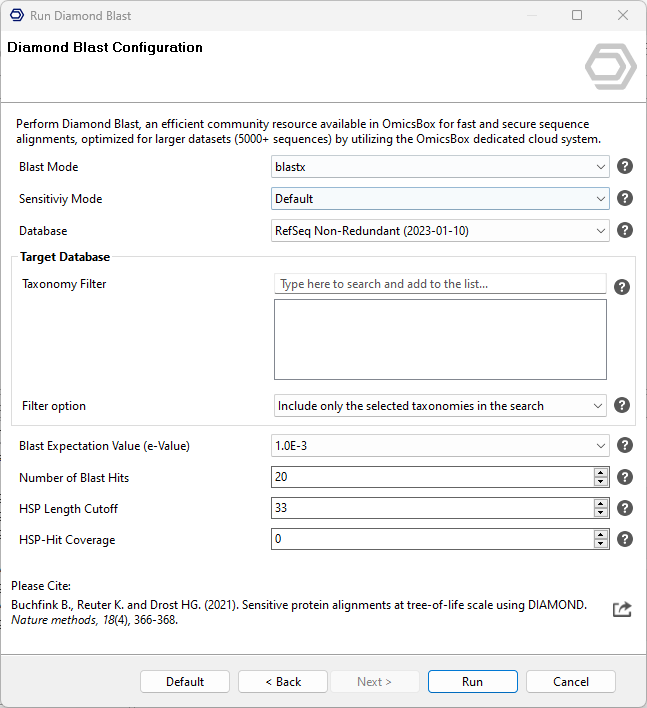
Figure 1. Diamond Blast Configuration page.
Custom Diamond database
A custom Diamond database can be uploaded and used for Diamond Blast.
Prerequisites:
- A custom Diamond database as a single
.dmndfile. - The database must be properly formatted for Diamond Blast. OmicsBox does not accept raw data or FASTA files for database uploads.
Upload the custom database:
- Open OmicsBox and click the Cloud Files tab on the left-hand side to access the cloud storage.
- On any desirec location, right-click, and select New Folder to create a folder.
- Drag and drop the
.dmndfile into the new folder.
Use the custom database: in the Diamond Blast configuration, open the Database drop-down and select the previously uploaded custom Diamond database. The custom database is automatically located within the cloud files by the .dmnd extension.
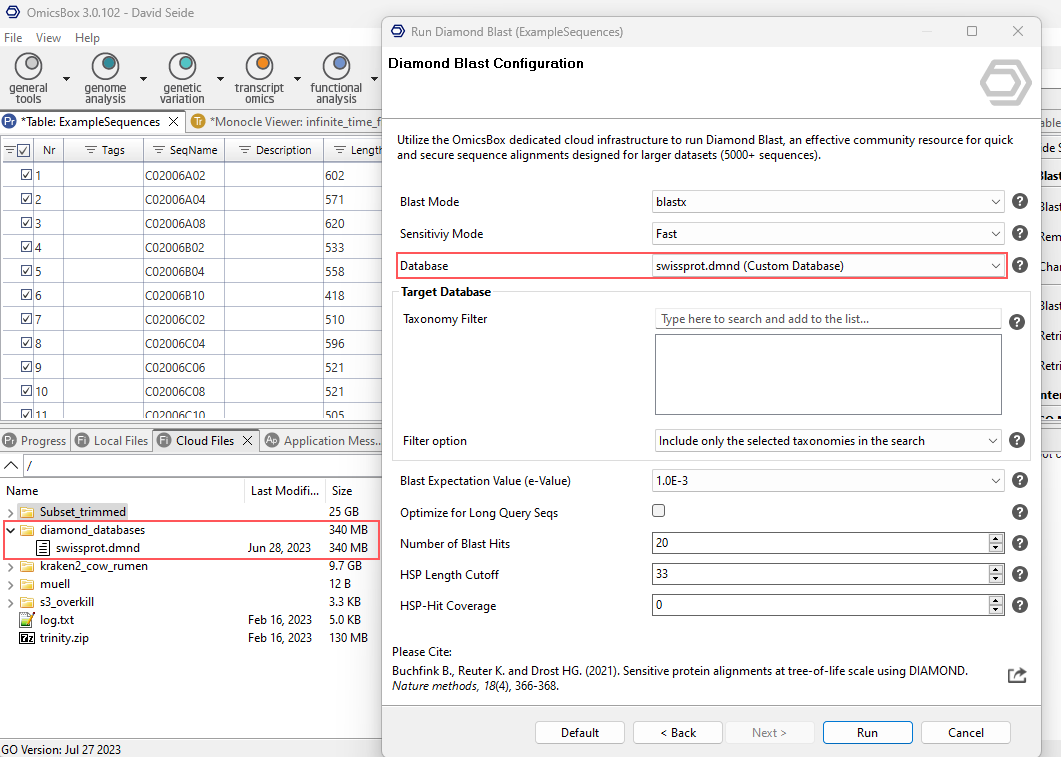
Figure 2. Selecting a custom database from the user's Cloud Files.
Results
As with any Blast search, sequences with hits turn orange and sequences without hits turn dark red in the functional annotation project. The Blast statistics charts, Blast side-panel actions, and the individual Blast result view are described in the Blast Search Results section.
OmicsBox Engine
This tool can be run from the command line via the OmicsBox Engine.
Command: omicsbox diamond [options]
Execution: Cloud (runs on OmicsCloud)
Input constraint: provide exactly one of:
--i-local-project,--i-input-fasta.
Inputs
| Flag | Type | Required | Description |
|---|---|---|---|
--i-local-project |
file | Yes | Sequence Project (.box) |
--i-input-fasta |
file | Yes | Query Input |
Parameters
| Flag | Type | Default | Range / Candidates | Required | Description |
|---|---|---|---|---|---|
--blast-mode |
enum | blastx | blastxblastp |
No | Blast Mode |
--long-queries |
boolean | false | No | Optimize for Long Query Seqs | |
--taxa-filter-option |
enum | included | includedexcluded |
No | Filter option |
--blast-expect-value |
enum | 1.0E-3 | 1000105 |
No | Blast Expectation Value (e-Value) |
--database |
enum | nr_20260702 | refseq_protein_20260702refseq_protein_20250925refseq_227_20241120 |
No | Database |
--number-of-hits |
integer | 20 | 1 – 100 | No | Number of Blast Hits |
--blast-min-hsplength |
integer | 33 | 1 – 5000 | No | HSP Length Cutoff |
--hsp-hit-coverage-cutoff |
integer | 0 | 0 – 100 | No | HSP-Hit Coverage |
--species |
string | No | Taxonomy Filter | ||
--sensitivity |
enum | more_sensitive | fastmid_sensitivestandardsensitivemore_sensitivevery_sensitiveultra_sensitive |
No | Sensitiviy Mode |
Parameter relationships
| Flag | When | Effect | Affected flags |
|---|---|---|---|
--blast-mode |
blastp |
disables | --long-queries |
--long-queries |
true |
disables | --number-of-hits |
Global options (
--local-folder,--cloud-folder,--output-format,--config,--detach,--verbose, …) are shared by every Engine tool and are not repeated here — see the OmicsBox Engine reference.