NCBI Blast - Local
Introduction
Local Blast searches the project sequences against a local database on the same machine. OmicsBox can build a Blast database from a FASTA file with the Create Blast Database tool. Databases must be formatted for NCBI Blast+.
Local Blast is one of the ways to run a Blast search in OmicsBox. See Blast Search for the overall Blast workflow and where to launch it.
Please cite NCBI for Local Blast and pre-formatted databases: https://www.ncbi.nlm.nih.gov/books/NBK569850/.
Run
Select NCBI Blast - Local on the first page of the Blast wizard. The wizard provides the following parameters:
-
Blast Program. The Blast algorithm, chosen according to the type of the query sequences and of the database. The main programs are:
blastp: protein query against a protein database.blastx: nucleotide query, translated in all six reading frames, against a protein database. Used to find the potential protein products of an unknown nucleotide sequence.blastn: nucleotide query against a nucleotide database.tblastn: protein query against a nucleotide database translated in all six reading frames.tblastx: nucleotide query, translated in all six frames, against a nucleotide database also translated in all six frames.
Several programs also offer variants:
-fast(for exampleblastx-fast,blastp-fast,tblastn-fast): a faster preset that uses a larger word size, best suited to finding closely related sequences at the cost of sensitivity for distant matches.-short(for exampleblastp-short,blastn-short): tuned for short query sequences, using a smaller word size and adjusted scoring.megablast(ablastnmode): the fastest nucleotide search, for highly similar sequences such as those within a species.dc-megablast(discontiguous megablast, ablastnmode): for more divergent nucleotide sequences, such as cross-species comparisons.
Only searches against a protein database (
blastp,blastx) produce hits usable for GO mapping and annotation, since Gene Ontology terms are linked to proteins. Default:blastx-fast. * Strand. Whether to search both strands, only the forward strand, or only the reverse strand. Available only forblastxandblastx-fast. * Formatted Blast Database. The local database to search against. Choose any file of a database formatted for NCBI Blast+ (.psq,.nsq,.pal, or.nal); the path must not contain spaces. Use Create Blast Database to build a database from a FASTA file. * Blast Expectation Value (E-Value). The statistical significance threshold for reporting matches. Lower values are more stringent.. * Number of Blast Hits. The number of alignments to retrieve (1-500). * Blast Description Annotator. Finds the best possible description for each sequence from its Blast result. * Word Size. The length of the initial word of the local alignment. Smaller values increase sensitivity; larger values increase speed. * Low Complexity Filter. Applies SEG filtering of low-complexity protein regions before the search. * Number of Threads. The number of CPU cores to use; more threads run faster. By default all available cores are used. * HSP Length Cutoff. The minimal length of the first HSP of a hit, used to exclude hits with only small local alignments. * Filter by Description. Removes hits whose description contains the given text (semicolon-separated terms). * Blast XML2 Folder. If checkec, the results are saved as XML2 files in the selected output folder.
See this tutorial on how to download NCBI pre-formatted databases.
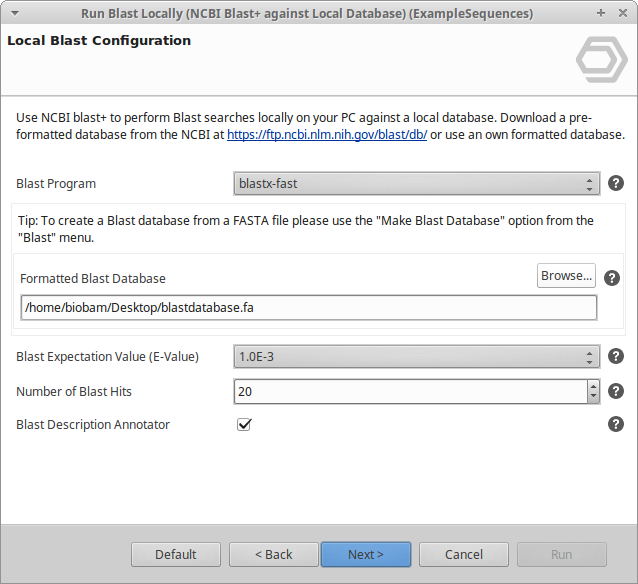
Figure 1. Local Blast Configuration page.
Results
Sequences with hits turn orange and sequences without hits turn dark red in the functional annotation project. The Blast statistics charts, Blast side-panel actions, and the individual Blast result view are described in the Blast Search Results section.